
|
Introduction
Interactions that are mediated by flexible peptides are abundant and play a key role in the protein-protein interaction network and specifically in signal transduction and regulation. These interaction are gaining much interest due to their cardinality, and their promising potential to provide new leads for drug targets. Peptides often lack a distinct fold in their unbound state, and go through simultaneous binding and folding upon encountering their target protein receptor. This is a challenging modeling task. Protocol outline The Rosetta FlexPepDock protocol for high-resolution docking of flexible peptides is outlined in Figure 1. It mainly consists of two alternating modules that optimize the peptide backbone and rigid body orientation, respectively, using the Monte-Carlo with Minimization approach. The starting structure is refined in 200 independent FlexPepDock simulations. 100 of the simulations are carried out strictly in high-resolution mode, while 100 of the simulations include a low-resolution pre-optimization step, followed by the high-resolution refinement. A total of 200 models are thus created and then ranked based on their Rosetta generic full-atom energy score. For more details, please see the method section of Raveh et al.. 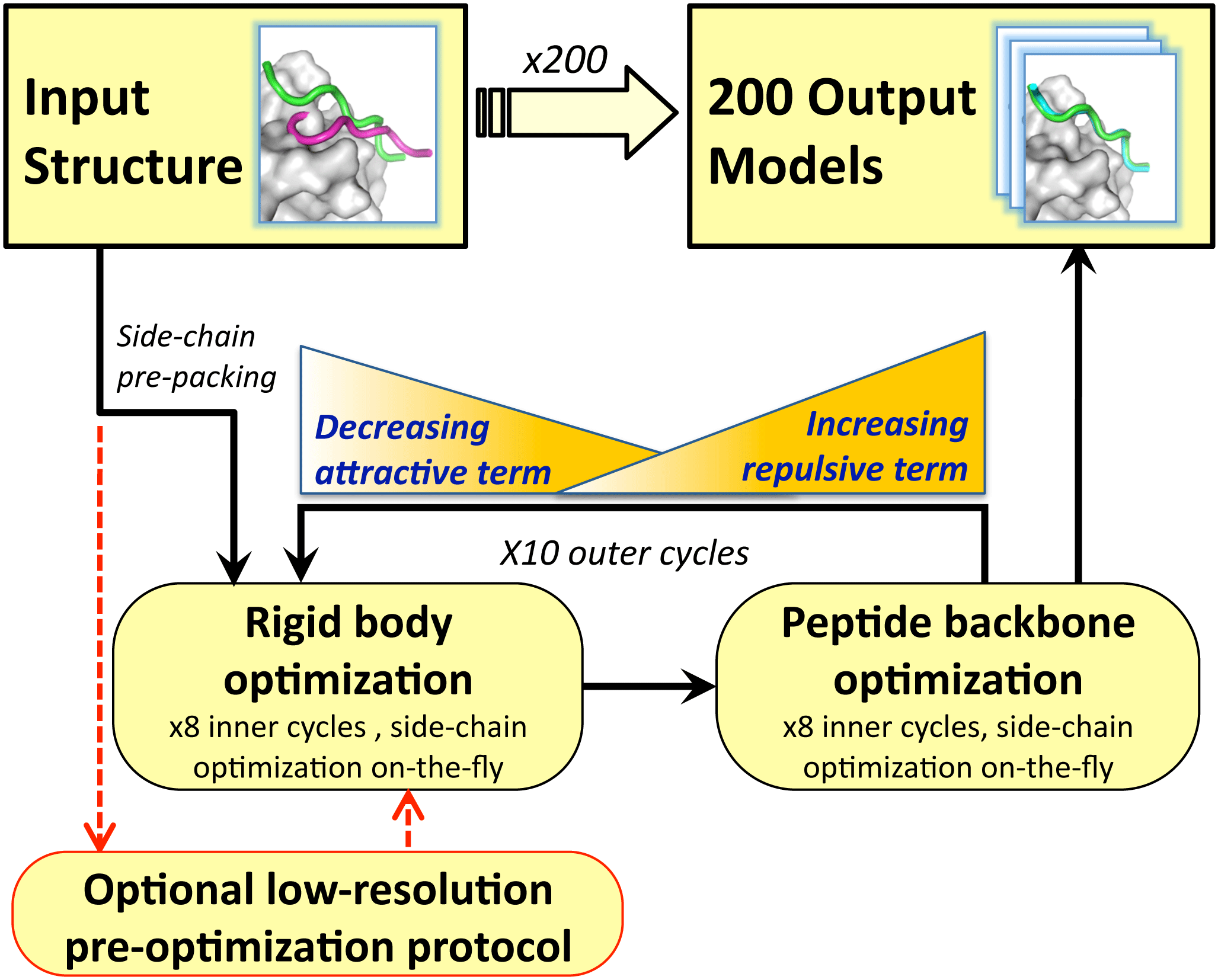
Figure 1. Outline of the FlexPepDock Protocol. Credit: Raveh et al. 2010, Proteins. Performance & Benchmarking FlexPepDock was thoroughly benchmarked against a set of perturbed peptide-protein complexes and an effective range of sampling was defined. For peptides with initial backbone (bb) RMSD of up to 5.5A, FlexPepDock is able to create near-native models (peptide bb-RMSD <2A) in 91% of the cases for the bound receptor, and rank them as one of the top 5 models in 78%. In the challenging task of unbound (apo) docking, near-native models were sampled in 85% of the cases and ranked correctly in 59% (for starting structures within 5.5A bb-RMSD from the native). The accuracy of the protocol for high-resolution modeling was tested on consecutive 4-mers, as peptide binding is often mediated by short, highly-conserved motifs. Indeed, for starting structures within 3.5A bb-RMSD, FlexPepDock managed to sample all-atom sub-angstrom (<1A) 4-mers for 82% of the bound cases and 62% of the unbound cases, and to rank them among the top five models in 62% and 35% of the cases, respectively. In cases where no information is available about the conformation of the peptide backbone, docking can be started from an extended conformation of the peptide. In a benchmark in which the peptide was docked starting from an ideal extended backbone conformation (+/- 135 deg. for all phi/psi angles) based on a single anchor residue, near-native solutions could be sampled in 66% of the 71 non-helical complexes (31% for sub-angstrom models), and ranked among the top five solutions in 49% of the cases (24% for sub-angstrom models). Fore more results and details, read Raveh et al.. For more details regarding technical operation of the server and analysis of results, visit the "Usage & FAQ" page. |